近几年,基因治疗的IND申报数量激增,其中AAV类产品因CMC质量数据不充分被打回的案例占比超过40%(据业内不完全统计)。很多团队在早期研究阶段只盯着”滴度够不够”,用qPCR测一个漂亮的vg/mL数字就信心满满地推进,却在IND审评阶段被追问:你的空壳率是多少?这个问题,足以让一个准备了两年的项目原地停摆。本文将直接告诉你,为什么”滴度”只是一张门票,而空壳率才是决定能否过关的底牌。
滴度的谎言,你听懂了吗?
滴度≠有效剂量
我们常说的”AAV滴度”,大多数时候指的是基因组滴度(Genomic Titer),即用qPCR或ddPCR检测到的含基因组的病毒颗粒数(vg/mL)。这个数字听起来很直观,但它有一个致命的前提假设——所有被计数的颗粒都是”满的”、有功能的载体,有效剂量就只包含被计算的病毒颗粒。
现实是,AAV在生产过程中会同时产生如下几种不同形式:
Full capsids(完整衣壳):含有完整治疗基因组,是你真正想要的东西
Empty capsids(空衣壳):蛋白外壳完整,但没有包裹目的基因
Partial capsids(部分包装衣壳):包裹了不完整或非目的DNA片段
Over-packaged capsids(过度包装衣壳):基因组多态性或过度包装了其它DNA
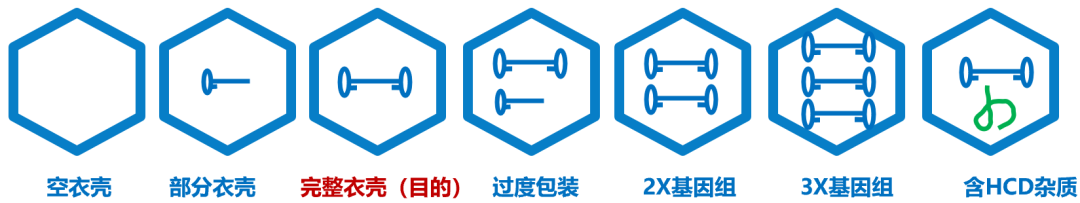
图1 各种形态的AAV病毒颗粒示意图
qPCR/ddPCR只计算有目的基因组的拷贝,因此空衣壳不会被纳入vg/mL的计数。但问题在于:在你给患者注射的那支注射液里,空衣壳可能占到总颗粒数的30%~50%[1],它们一样会被免疫系统识别,会占据受体结合位点,从而引发肝毒性和炎症。同时,如果包装的基因组不完整,基因组滴度可能就是个虚值,无法反映真实的有效的全长基因组情况。
用一句话总结:高滴度的背后,可能藏着大量的”空炮弹”或“残次品”,并非全部有效剂量。
空壳率分析
空壳率(Empty Capsid Ratio)——你注射的究竟是药还是”壳”?
空壳率的检测方法决定了数据的可信度。目前主流的空壳率检测方法有五种,准确性和适用场景各有不同:

表1 主流的空壳率检测方法
行业现状警示:部分CRO/CDMO提供的检测报告仅包含A260/A280数据,而将其包装成”全面的质量检测报告”。如果你的供应商无法提供AUC数据,那这份质量报告在IND审评面前基本等同于废纸。
空壳率的监管标准在哪里?
FDA和EMA的指南均要求对AAV空壳率进行监测和报告[2,3]。2022年,Dark Horse Consulting向FDA提交了建议性草案,提出最大放行标准应≤30%空壳率,但该标准尚未被FDA正式采纳为强制性法规[4]。目前行业内GMP产品的空壳率控制目标多在10-30%范围内,具体取决于产品和适应症。
决策建议:
- 科研阶段:要求供应商至少提供TEM+AUC双重验证数据
- IND准备阶段:推荐以AUC作为放行检验方法,或开发SEC-MALS、Samux-MP等方法进行放行检验,拒绝接受仅有A260/A280的报告
- GMP阶段:空壳率应纳入质量放行标准(Specification),而非只是“informational”数据
代表性案例:一个因”假滴度”差点毁掉IND的真实故事
某基因治疗团队,在完成动物实验后信心十足地准备递交IND。其中,动物实验用药的基因组滴度为1×10¹³ vg/mL,药效显著。
然而在进行IND前最后一轮质量核查时,委托第三方用AUC检测了空壳率——结果是62%。
这意味着什么?实际能发挥治疗作用的Full capsids滴度仅约为3.8×10¹² vg/mL,比预期的基因组滴度低了将近三分之二。更关键的是,当他们按照动物实验的剂量方案(基于vg计算)去换算人体剂量时,患者实际接受的”无效颗粒”负担远超预期,这在AAV2这一已知具有较强免疫原性的血清型上,是明显的安全隐患。
团队最终不得不重新优化纯化工艺(引入碘克沙醇密度梯度超速离心),将空壳率降至8%,再完成三批次连续的中试生产,整体IND准备周期延误了将近11个月。
关键教训:这个团队的滴度从未”说谎”,但他们从未问过那个正确的问题——这1×10¹³个颗粒里,有多少是真的有用的?
快速决策参考指南
- 如果你处于科研阶段(体外/小动物实验),供应商只提供qPCR滴度数据,那么可暂时接受,但要求同时提供TEM图像,并在动物实验设计中加入高/低剂量组,为后期空壳率校正预留数据空间。
- 如果你计划在12个月内递交IND,那么建议您寻找CDMO供应商启动CMC工作,并在最早期进行CMC成药性评估,尽量在早期对接使用AUC/SEC-MALS等方法表征AAV空壳率情况,对基因组完整性等进行评估考察,再有针对性的开展工艺开发和后续生产,积累多批次数据;
- 如果供应商告诉你”我们的空壳率很低”但无法提供AUC原始数据,那么这是一个红线信号(Red Flag),不要接受口头承诺,要求AUC原始数据文件或换供应商。
常见问题(FAQ)
Q1:ddPCR和qPCR测出来的滴度差别很大,以哪个为准?
两者检测原理相似,但ddPCR不依赖标准曲线,准确性和重复性更优,是CDE认可的检测方法。研究表明同一样品ddPCR与qPCR的测定值差异可达2-5倍[5]。IND申报推荐以ddPCR为主要方法,并在方法学验证(Method Validation)中提供两种方法的相关性数据。如果你的早期数据全部来自qPCR,切换到ddPCR后滴度可能发生变化,需要IND申报时提前做好剂量换算并进行充分说明。
Q2:空壳率高一点,能不能通过提高给药剂量来补偿?
不能这样操作,至少在人体试验设计中不能这样简单处理。空衣壳不是”惰性”物质,它们会:①激活先天免疫应答,消耗中和抗体容量;②与全衣壳竞争受体结合位点,降低转导效率;③增加肝脏和目标组织的总颗粒负担,放大脱靶毒性风险。简单提高剂量等于在放大所有已知和未知的风险,这在IND审评中是无法被接受的逻辑。
作者按:这篇文章里的每一个”坑”,都是行业里真实踩过的。如果你觉得有用,转给你们团队的CMC负责人看看,说不定能省下半年的时间。
参考资料
[4]https://www.darkhorseconsultinggroup.com/post/dark-horse-to-offer-proposed-fda-guidance-on-fte-capsid-ratio
[5] Lock M, Alvira MR, Chen SJ, Wilson JM. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum Gene Ther Methods. 2014 Apr;25(2):115-25.
本文由派真生物CMC技术团队整理,主要撰稿人:工艺开发团队。
关于派真
作为一家专注于AAV 技术十余年,深耕基因治疗领域的CRO&CDMO,派真生物可提供从载体设计、构建到 AAV、慢病毒和 mRNA 服务的一站式解决方案。凭借深厚的技术实力、卓越的运营管理和高标准的服务交付,我们为全球客户提供一站式CMC解决方案,包括从早期概念验证、成药性评估到IIT、IND及BLA的各个阶段。
凭借我们独立知识产权的π-alphaTM 293 细胞AAV高产技术平台,我们能将AAV产量提高多至10倍,每批次产量可达1×10¹⁷vg,以满足多样化的商业化和临床项目需求。此外,我们定制化的mRNA和脂质纳米颗粒(LNP)产品及服务覆盖药物和疫苗开发的各个阶段,从研发到符合GMP的生产,提供端到端的一站式解决方案。

相关服务
-
AAV病毒包装
- 6天极致交付
- 100+ 血清型,60+ 质量控制检测
- 已交付 50,000+ AAV,可放大至 GMP 级别
-
AAV病毒包装 – 科研级
- 6天极致交付
- 100+ 血清型,60+ 质量控制检测
- 已交付 50,000+ AAV,可放大至 GMP 级别
-
AAV病毒包装 – NHP级
- 严控内毒素,防污染管控
- ddPCR 精准定量,基因组完整性验证
- 卓越的一致性,安全性与有效性双重提升
-
AAV病毒包装 – HT级
- 最快 7 个工作日极速交付
- 采用 qPCR 滴度测定,具备高性价比
- 优化工艺以在细胞培养中实现卓越性能
-
AAV分析检测服务
- 全面的 AAV 检测:滴度、纯度、安全性等一应俱全
- 结果快速可靠,专家全程指导
-
AAV血清型筛选
- 定向进化 + 理性设计,精准打造理想衣壳
- 体内数据可信赖,候选分子轻松筛选
- 项目专属保密,突变体逐一验证
-
AAV现货
- 现货直发,到手即用
- 品类齐全,覆盖多元化科研需求
- 严苛质控,品质稳定可靠
-
AAV载体设计与构建
- 定制化AAV质粒,实现高效基因递送
- 通过piVector轻松设计,集成特定应用所需元件
- 支持CRISPR、shRNA及其他类型的AAV载体