

中枢神经系统疾病的基因治疗:方式、传递和转化挑战
研究概述
2024年6月19日,哈佛医学院的Nitin Joshi教授及其团队在Nature Reviews Neuroscience期刊发表了题为“Gene therapy for CNS disorders: modalities, delivery and translational challenges”的综述论文。该研究主要概述了针对中枢神经系统(CNS)疾病的基因治疗策略,包括基因治疗的不同模式、在CNS中的传递策略,以及临床转化过程中遇到的挑战。研究团队强调了不同传递方法和给药途径之间的相互联系,并探讨了如何克服这些挑战,以提高基因治疗在CNS疾病治疗中的临床应用性和可行性。
正文
1. 引言
发现导致中枢神经系统(CNS)疾病的遗传因素为基因治疗铺平了道路,这为这些病理的药物开发提供了革命性的潜力。与传统方法不同——例如小分子、单克隆抗体和蛋白质治疗——基因治疗针对疾病的潜在遗传原因,提供了改进的特异性和治疗效果,以及个性化的治疗选择。已经开发了各种工具,如DNA质粒、成簇的规则间隔的短回文重复序列(CRISPR)相关的核酸酶(Cas核酸酶)和RNA干扰(RNAi),分别促进体内基因增强、编辑和沉默。这个工具箱通过新基因操作技术的发现和系统的进展而得到丰富,这些系统使得安全有效地传递基因治疗成为可能。
截至2023年,已有110种基因治疗干预措施进入临床试验,用于治疗CNS疾病(见补充表1)。其中四种(nusinersen(Spinraza)、onasemnogene abeparvovec(Zolgensma)、elivaldogene autotemcel(Skysona)和tofersen(Qalsody))已获得美国食品药品监督管理局(FDA)的批准,还有一种(eadocagene exuparvovec(Upstaza))已获得欧洲药品管理局(EMA)的批准。尽管这些成功推动了CNS疾病的基因治疗进展,但这种方法的全部潜力受到两个主要挑战的阻碍:血脑屏障(BBB),它阻止了系统性给药分子进入大脑;以及大脑复杂的解剖结构,治疗剂必须导航到达特定区域或细胞类型。由于其高转导效率,腺相关病毒(AAVs)已在大多数临床试验中用于传递CNS基因治疗。然而,大多数基于AAV的方法依赖于侵入性的局部给药途径。非病毒载体比AAVs表现出更低的免疫原性和更大的包装能力,但面临BBB穿透和转染效率的挑战。替代的给药途径或BBB渗透方法也有其自身的局限性。
CNS基因治疗的转化成功依赖于治疗方式、传递工具和给药途径之间复杂的相互依赖性。在这篇综述中,作者探讨了这些变量及其相互联系如何影响临床适用性、可行性和可转化性。作者首先提供了一个简明的基因操作技术概述,这些技术已被用于治疗不同的CNS疾病,并讨论了它们所呈现的独特机会和挑战。然后,作者概述了当前最先进的基因传递方法,总结了它们的进步和相关的问题。作者特别关注给药途径的重要性及其对剂量、生物分布、基因毒性和免疫原性的影响。此外,作者提供了涉及CNS疾病基因治疗的临床试验的现状,并讨论了从过去的不成功试验中学到的教训。最后,作者概述了未来的发展方向和潜在的进步,这些可能为开发临床上可转化的CNS疾病的基因治疗铺平道路。
2. CNS疾病的基因治疗方式
CNS基因治疗策略大致可以分为三种类型。基因增强为那些稳态受到破坏的细胞提供蛋白质编码基因的功能性副本。基因组编辑涉及通过工程核酸酶在生物体DNA中插入、删除或改变目标序列。基因沉默不是直接影响感兴趣的基因,而是通过针对其转录产物来抑制其表达。
2.1 基因增强
基因增强包括基因替代和基因补充。基因替代通常涉及传递一个突变基因的校正版本,最适合治疗单基因疾病(图1),例如脊髓性肌肉萎缩症(SMA)。SMA的特征是SMN1基因的突变或缺失(或在某些情况下,其转化为SMN2),这导致生存运动神经元蛋白(SMN1)的表达下降,最终导致神经退行性变。FDA批准的基因治疗药物Zolgensma通过AAV9载体传递SMN1的功能性副本。在系统性给药后,Zolgensma穿过BBB并被运动神经元摄取。然后AAV9载体将遗传有效载荷释放到细胞核中,实现SMN1的稳定和持续表达。
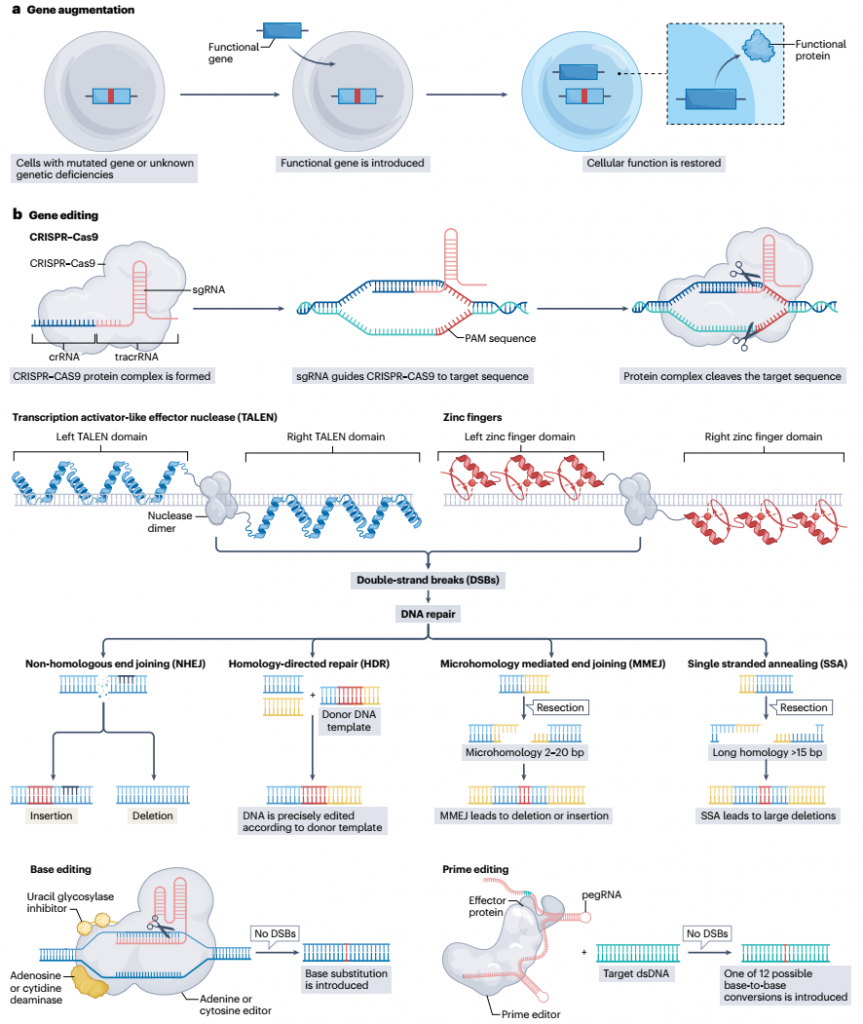
图1. 基因增强和基因编辑策略
Skysona是一种FDA批准的基因增强产品,可以延缓由于肾上腺脑白质营养不良引起的神经功能下降(一种由ABCD1编码的过氧化物酶体膜转运蛋白缺乏引起的疾病)的4至17岁男孩。Skysona是一种单剂量、个性化的基因治疗,使用慢病毒载体将ABCD1的功能性副本转染到来自肾上腺脑白质营养不良患者的造血干细胞中。然后,这些干细胞通过静脉输注回输给同一患者以治疗疾病。Upstaza是一种携带AADC基因(编码参与神经递质合成的酶)的重组AAV2载体,是治疗AADC缺乏症的单剂量基因增强疗法,已获得EMA的批准。Upstaza被颅内输送给患有这种疾病的儿童,并已显示出在恢复运动功能方面的前景。
尽管这些商业上成功的罕见例子,但许多基因增强产品在临床试验中表现出失败。例如,CERE-110(Ceregene Inc.)使用编码神经生长因子(NGF)的AAV2载体,旨在挽救与阿尔茨海默病相关的胆碱能功能障碍。尽管I期临床试验结果证明了干预措施的安全性、耐受性和长期生物活性表达,但II期试验没有产生有效结果。
为了确保复杂疾病基因增强治疗的成功临床转化,可能很重要的是要确定一个在疾病发病机制中起主要作用的目标基因。然而,必须考虑到CNS疾病的单基因遗传模式只占有限的患者群体。例如,有几种涉及致病基因的帕金森病的单基因形式。尽管这些单基因形式相对罕见,因此替换这些基因不适用于大多数帕金森病形式,但它们确实共同占到了家族性帕金森病的约30%和散发性帕金森病病例的3-5%。同样,在阿尔茨海默病中,涉及单基因形式的疾病包括β-淀粉样前体蛋白(APP)、早老素1(PSEN1)和PSEN2的突变,值得积极考虑作为靶点。例如,APP突变出现在2-18%的患有常染色体显性早发性家族性阿尔茨海默病患者中,PSEN1突变影响了18-55%的家族性阿尔茨海默病患者,而PSEN2突变迄今只影响了极少数个体。总的来说,阿尔茨海默病的单基因形式可能只占所有阿尔茨海默病病例的一小部分(<5%)。尽管如此,制定临床试验设计时仍需考虑这些稀少的单基因遗传模式。事实上,最近的一项研究报告称,将编码野生型PSEN1的AAV9载体输送到患有单基因常染色体显性阿尔茨海默病的非人灵长类动物中,成功地恢复了皮层和海马中的PSEN1水平。同样,在小鼠和基于患者的细胞模型中,AAV-PSEN1载体显示出满意的结果。
基因补充,是基因增强的另一种形式,涉及在不总是涉及遗传突变的疾病个体中提供基因的功能性副本。这种方法的一个例子是ProSavin(最初由Oxford BioMedica开发,授权给Sio Gene Therapies(Sio),以前称为Axovant)——一种三顺反子慢病毒载体,携带三个编码多巴胺合成酶的基因。ProSavin被测试其恢复帕金森病患者多巴胺水平的能力,并是第一个进入CNS疾病治疗临床试验的基于慢病毒的基因疗法之一。在I/II期开放标签试验中,ProSavin对晚期帕金森病患者安全且耐受性良好,从基线到治疗后1年内,运动行为有显著改善。5年后的长期随访显示,ProSavin继续安全且耐受性良好,一些患者的运动行为有中等改善。然而,关于ProSavin管理后长期持久性和在其他临床结果中的有效性的关注部分归因于传递和载体设计的问题,这促使开发了OXB-102载体。OXB-102.一种改进的基因表达盒,在非人灵长类动物中显示出有希望的临床前疗效。然而,由于开发这种治疗的公司解散,OXB-102的I/II期临床评估被终止。在帕金森病中探索的其他基因补充靶点,如AADC、neurturin(NTN)、谷氨酸脱羧酶(GAD)和胶质细胞系衍生的神经营养因子(GDNF),也未能进入III期试验。传递工具的限制、给药途径、剂量方案和基因转导效率,以及对帕金森病对基因传递和表达的病理生理效应了解不足,被认为是这些失败的可能原因。
总体而言,基因增强和补充的潜在优势在于它们能够以最小的剂量实现治疗效果(为使用相对侵入性的给药途径提供了机会)。然而,其功效取决于传递工具促进基因稳定整合到相关细胞的能力。
2.2 基因编辑
与基因替代相比,用于CNS(中枢神经系统)疾病的基因编辑仍处于探索阶段。用于基因编辑的三大核酸酶家族是锌指核酸酶(ZFNs)、转录激活因子效应子核酸酶(TALENs)和Cas核酸酶。图1和方框1提供了每种方式实现基因编辑的机制概要。
这些核酸酶实现编辑的不同机制对它们的设计标准有影响,这决定了它们的整体效用和适用性。例如,与TALENs和Cas核酸酶相比,ZFNs的体积更小,使它们更容易被输送到大脑,但设计ZFNs需要比生产TALENs更复杂的蛋白工程。此外,与TALENs和Cas核酸酶相比,ZFNs更有可能产生非目标效应。然而,研究已经展示了新ZFN架构实现精准基因组编辑的潜力,在黏多糖贮积症I/II型和血友病B患者中应用,并可能为神经系统疾病开辟新的可能性。与ZFNs和TALENs相比,CRISPR–Cas9系统提供了一个更简单、更稳健的方法,它只需要一个单向导RNA(sgRNA)和一个Cas9核酸酶来分别结合和切割目标DNA序列。CRISPR–Cas9系统已经被用于各种临床前和临床应用,并且针对CNS疾病的几个临床试验正在进行中。尽管CRISPR干扰(CRISPRi)仍然是主要技术,CRISPR激活(CRISPRa)是一种较新的策略,其中效应分子缺乏内切核酸酶活性,但引导RNA或非活性Cas核酸酶具有可以诱导基因表达的转录激活元素。这种方法主要在临床前研究中进行了探索,并在神经系统疾病的背景下显示出前景。例如,CRISPRa介导的Kcna1(编码钾通道亚单位)上调,最小化了癫痫动物模型中的癫痫发作和认知障碍。尽管CRISPR–Cas9比其他核酸酶家族具有几个优点,例如使用方便、能够适应任何基因组区域的目标选择、高效的位点选择能力和同时靶向多个位点(多重)的能力,但它也容易受到非目标效应的影响,这需要仔细设计引导RNA。
所有三种核酸酶通过在感兴趣的基因组位点引入双链断裂(DSBs),触发内源性修复机制的招募,该机制可以通过四种主要途径在切割位点插入、替换或删除核苷酸:非同源末端连接(NHEJ)、同源导向修复(HDR)、单链退火(SSA)和微同源介导的末端连接(MMEJ;一种替代末端连接的形式)。NHEJ是一个容易出错的途径,它依赖于催化切割产生的钝端的简单连接,而HDR需要一个供体DNA模板,允许高保真度修复。SSA和MMEJ也通过连接断裂附近的同源序列或通过退火暴露的微同源重复序列实现快速但突变的修复。NHEJ是核酸酶切割后的主要修复过程,但多种因素(如细胞周期阶段、对损伤的转录程序反应、局部染色质相互作用和切割位点相邻序列的身份)决定最终选择哪种修复过程。考虑到NHEJ的总体保真度较低,可能会产生有害产物,正在开发几种基于CRISPR的工程方法,以抑制NHEJ途径或优先招募并提高在修复位点更准确的HDR的效率。
较新的基因编辑技术,如碱基编辑和先导编辑,与基于核酸酶的方法相比提供了更大的精确性,因为它们在不引入双链断裂的情况下改变核苷酸序列。碱基编辑允许通过胞嘧啶碱基编辑器控制C•G到T•A的转换,以及通过腺嘌呤碱基编辑器实现A•T到G•C的转换。先导编辑扩大了可能编辑的范围,理论上可以实现所有12种可能的碱基转换(包括转换和颠换),而无需供体DNA模板或双链断裂,从而最小化了对基因组的附带损害风险。先导编辑和碱基编辑已经建立的能力,可以恢复线粒体DNA突变后的线粒体功能,为这些工具应用于涉及线粒体失衡的神经条件提供了可能性。尽管先导编辑尚未在神经退行性模型中证明成功,但碱基编辑已被用于在肌萎缩侧索硬化症(ALS)小鼠模型中纠正驱动该疾病的SOD1基因突变。除了像ALS这样的疾病需要线粒体恢复外,碱基编辑也显示出在SMA模型中将有缺陷的SMN2转换为健康的SMN1的前景。
基因编辑导致DNA的永久性变化,通常在特定的内源位点,这意味着只需要一次干预。这就证明了使用侵入性程序(如立体定向手术和脑室内及脑实质内注射)的合理性。此外,立体定向脑实质内注射可用于空间靶向的深脑基因疗法。然而,评估应用编辑的疾病阶段也至关重要。以快速空间进展为特征的神经退行性疾病可能受益于能够使工程核酸酶在大脑中扩散的策略,如脑室内和脊髓腔内注射。然而,这些给药途径也可能导致基因疗法的系统吸收,因此重要的是要设计最小化这种风险的传递载体。英国药品和保健品监管署(MHRA)和FDA最近批准了世界上第一种CRISPR疗法exagamglogene autotemcel(CASGEVY),该疗法利用电穿孔介导的体外CRISPR编辑造血干细胞来治疗镰状细胞病。作者相信,CRISPR基因编辑技术在CNS疾病中的应用也将很快进入临床评估。
2.3 基因沉默
CNS基因沉默工具中最常用的是RNA干扰(RNAi)、反义寡核苷酸(ASOs)、CRISPRi和催化核酸(CNAs)(图2)。其中,只有CRISPRi直接针对病理性DNA序列来控制基因表达;其他方法通过针对它们的mRNA转录本来沉默基因。与增强和编辑不同,基因沉默的持续时间通常很短,从几天到几个月不等(取决于所选择的模式和传递载体)。因此,需要多次剂量来提高CNS疾病患者的生活质量和生存率。
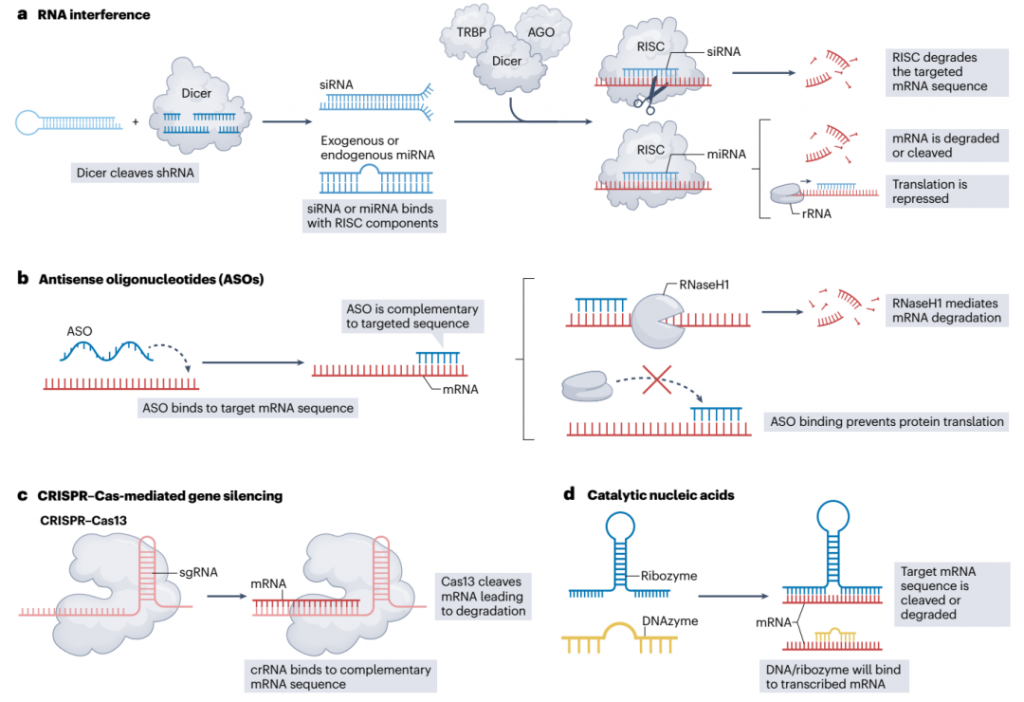
图2. 基因沉默模式
RNAi是一种内源性调控过程,依赖于短发夹RNA(shRNA)、小干扰RNA(siRNA)或微小RNA(miRNA)通过RNA诱导的沉默复合体(RISC)诱导特异性基因表达抑制。尽管已有五种FDA批准的siRNA药物用于不同的疾病,但siRNA在CNS中的直接应用很少见。
迄今为止,唯一进入CNS疾病临床试验的siRNA治疗是ALN-APP(Alnylam Pharmaceuticals公司),这是一种针对APP mRNA的合成siRNA。ALN-APP实现了有效的CNS传递,在啮齿类动物和非人灵长类动物中实现了长达3-4个月的基因沉默,经过一次脊髓腔内注射。在阿尔茨海默病患者的I期研究中,ALN-APP被证明是安全且耐受性良好的,并且降低了脑脊液中的淀粉样β蛋白、可溶性APPα(sAPPα)和sAPPβ(与阿尔茨海默病相关的APP蛋白水解产物)的水平。另一种方法是利用慢病毒传递针对BACE1(编码参与APP蛋白水解的酶)的siRNA,有效地降低了海马体中的淀粉样β蛋白水平,并减少了阿尔茨海默病动物模型中的淀粉样β蛋白斑块积累。其他研究已经针对与阿尔茨海默病相关的各种基因,包括APP、微管相关蛋白tau(MAPT)、电压依赖性阴离子通道1(VDAC1)、与Rho相关的卷曲螺旋含有蛋白激酶2(ROCK II)、PSEN1和乙酰辅酶A乙酰转移酶(ACAT1)。siRNA也已在动物模型中用于针对帕金森病中的α-突触核蛋白(SNCA)、亨廷顿病中的亨廷顿蛋白(HTT)以及与多发性硬化症相关的各种基因,以及用于治疗脊髓损伤。
miRNAs的失调与各种神经系统疾病的发病机制有关,这表明基于miRNA的治疗可能通过调节目标基因的表达来针对其他无法治愈的CNS疾病。例如,miR-SOD1.使用AAV系统性传递,在ALS小鼠模型中延长了生存期,并在非人灵长类动物模型中降低了脊髓SOD1水平。鉴于这一初步的临床前成功,两名ALS患者接受了脊髓腔内注射AAV-miR-SOD1.第一名患者的一条腿的力量有所改善,而第二名患者在ALS功能的综合测量上保持了稳定分数,没有观察到其他临床益处。研究不能得出结论,即SOD1本身的抑制影响了第一名患者的临床过程,因为脑脊液中的SOD1水平没有受到影响。AMT-130(UniQure公司)的I/II期试验,一种AAV编码针对早期亨廷顿病的HTT的miRNA,表明治疗被良好耐受,并成功抑制了突变HTT(mHTT),导致脑脊液中神经丝轻链(NfL)(神经退行性的指标)水平的降低。同样,正在进行一项I/IIa期试验,以评估AMT-260(UniQure公司)在单侧难治性内侧颞叶癫痫成人患者中的应用。AMT-260使用miRNA靶向GRIK2(编码一种谷氨酸受体亚单位),并通过磁共振成像引导的对流增强传递进行管理。
ASOs通过招募RNAse H或其他核酸酶降解mRNA,或者简单地阻碍核糖体接触到mRNA来降低基因表达。一些ASOs通过结合mRNA的剪接增强区域,抑制mRNA剪接抑制,或者通过结合剪接沉默子区域,引起剪接激活和降解。Nusinersen,一种FDA批准用于治疗SMA的ASO,展示了后一种方法的成功。Nusinersen修改了SMN2前mRNA剪接,导致功能性SMN2的产生,并具有主要归因于存在ribose-O-(2-methoxyethyl)修饰和磷酸硫代骨架,增加了其稳定性的长期消除半衰期。在milasen中也使用了类似的化学修饰策略,milasen是一种22个核苷酸的ASO,为神经元脑白质营养不良7型患者提供了个性化治疗。Milasen针对MFSD8前mRNA的隐秘剪接位点,导致癫痫发作显著减少,并为开发患者定制的ASO治疗提供了模板。
针对其他神经系统疾病的ASO开发也显示出相当的前景。Tofersen已证明在降低SOD1蛋白的CSF水平方面有效,推动其进入III期试验,并因SOD1突变引起的ALS获得FDA的加速批准。其他ALS靶点,如FUS、C9ORF72和ATXN2.目前正在ASO治疗的临床试验中探索。正在进行一项I/II期临床试验,以评估ASOs降解MAPT mRNA以减少早期发病阿尔茨海默病的tauopathy负担。Tominersen旨在针对亨廷顿病中的mHTT,尽管最初的III期试验因风险-效益比差而暂停,但最近的事后分析导致了II期剂量寻找试验的重新启动,以调查亨廷顿病负担较低的个体是否可能从tominersen治疗中受益。其他针对mHTT表达的ASOs的试验,如WVE-120101和WVE-120102(Wave Life Sciences),因缺乏疗效而在IIb/IIa期终止。尽管遇到了一些挫折,但ASOs的未来看起来很光明:Ionis Pharmaceuticals公司的反义药物管线是健全的,并且适用于各种神经系统疾病。
CRISPRi通过一种核酸酶失活的Cas蛋白,dCas9.实现翻译控制,该蛋白能够将抑制域招募到基因的转录起始位点。CRISPRi已被用作研究工具,以研究疾病变体的机制、治疗靶点、基因表达的因果变化以及细胞类型选择性脆弱性决定因素,并且也已被用于神经疾病的治疗靶向。例如,在亨廷顿病的小鼠模型中,将Cas9或dCas9与包裹在慢病毒中的sgRNA结合后注射到纹状体,显著抑制了mHTT达4周,导致神经保护和改善行为功能。通过AAV载体将RfxCas13d(来自Ruminococcus flavefaciens的Cas13核酸酶)传递到SOD1的目标上,可在ALS的小鼠模型中减少缺陷基因表达,延缓疾病进展并延长生存期。CRISPR–Cas13也被用于针对亨廷顿病模型中的mHTT mRNA,导致HTT蛋白及其聚集物减少。类似地,一种旨在针对共济失调蛋白2的高保真度RfxCas13d核酸酶在ALS和小脑变性病的小鼠模型中减轻了TDP43病理。研究人员还针对亨廷顿病的啮齿类和人类模型中的mHTT RNA设计了一个RfxCas13d系统。编码此Cas13d系统的AAV载体在小鼠纹状体内部传递,导致mHTT聚集物下调、纹状体萎缩减少和运动协调性改善。另一项研究使用催化非活性Cas蛋白CasRx,针对pre-mRNA进行选择性剪接,缓解了额颞叶变性疾病神经模型中失调的tau异构体比例。
值得注意的是,CRISPRi介导的沉默持续时间取决于其组分的持久性和细胞内目标RNA和蛋白质分子的周转率。
催化核酸(CNAs),如核糖酶和脱氧核酶,为基因沉默提供了替代途径。核糖酶修改mRNA剪接并降解mRNA,而脱氧核酶在特定位点识别并切割mRNA。合成脱氧核酶已成功在亨廷顿病的小鼠模型中沉默mHTT表达,降低细胞毒性和神经毒性。核糖酶和脱氧核酶也已被用于针对阿尔茨海默病模型中的BACE1 mRNA。类似地,通过重组AAV载体传递至黑质的α-突触核蛋白核糖酶在帕金森病的大鼠模型中保护神经元免受凋亡死亡,并延长了治疗大鼠的存活期。
3. 在CNS中的基因传递策略
识别有缺陷的基因和纠正这些基因的工具只是探索治疗途径的一部分。精心构建和优化传递系统以及选择合适的传递途径对于实现理想效果的同时最小化细胞毒性至关重要(图3)。
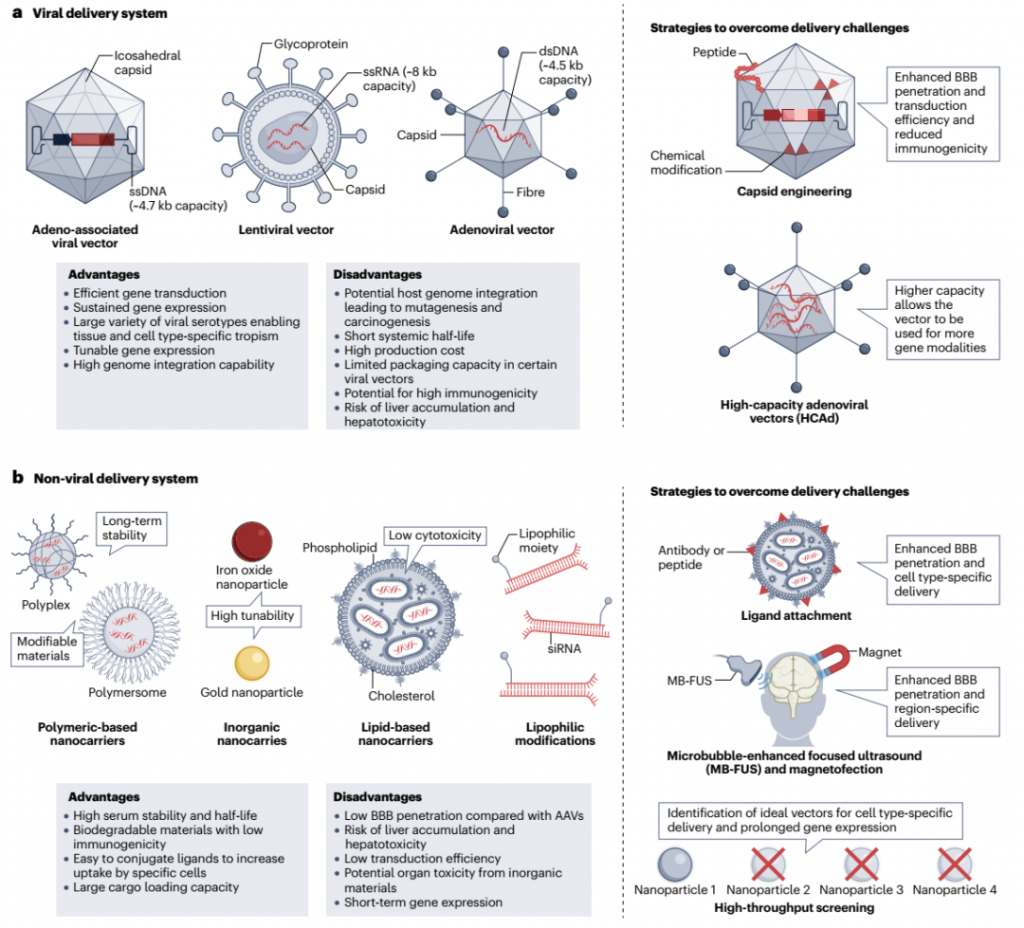
图3. 病毒和非病毒传递系统的比较
3.1 病毒传递
病毒用于基因治疗的使用历史悠久,最早成功的临床治疗可追溯至1985年。该策略涉及将病毒基因组替换为定制的载体,并利用病毒固有的能力劫持宿主细胞的分子机制,以整合和复制治疗基因。由于它们对神经细胞的强烈转导、高转基因表达和对工程策略的适应性,各种病毒载体已被用于开发CNS基因治疗。
慢病毒载体在传递功能基因治疗脑白质营养不良方面显示出前景。例如,Skysona向患有脑肾上腺白质营养不良的个体传递功能性ABCD1基因,而其他慢病毒载体已被用于向患有异染性脑白质营养不良的个体传递功能性的硫酸酯酶A(ARSA)基因。然而,慢病毒使用的整合机制有可能导致突变和致癌,因此目前仅限于在移植前对体外自体干细胞进行转染。尽管这种策略对特定病理有效,但在调控基因表达、系统性评估移植细胞、大脑分布和制造可扩展性方面面临挑战。
与慢病毒不同,AAV将其遗传物质传递到目标细胞而不整合到宿主的DNA中,最小化了突变风险。AAV还可以通过改变它们的血清型、伪型和衣壳蛋白来工程化,以展示更广泛的组织趋向性。尽管AAV9是首个在静脉注射后显著转导大脑的血清型,但AAVrh8和AAVrh10也被证明与AAV9同样有效。此外,AAVrh10在静脉注射后在脊髓中表达,AAV5有效地转导了光感受器和视网膜色素上皮细胞。不同的AAV血清型具有不同的CNS趋向性能力,但AAV9仍然是最被充分表征的血清型,并且最常被用于进一步工程化,以增强其穿透BBB的能力,并使其能够在脑实质中的神经元和非神经元群体中转导。
例如,一种AAV衣壳变体AAV.CAP-B10最近被证明能够以高度特异性向CNS神经元传递基因疗法。AAV衣壳也已被修改以展示细胞穿透肽,导致在小鼠和非人灵长类动物中CNS转导效率提高。同样,称为基于Cre重组的AAV靶向进化(CREATE)的衣壳选择平台已被用于识别比野生型AAV9更具有特异性和效率地转导CNS或外周神经系统的AAV9变体。更令人印象深刻的是,变体被工程化以在多种非人灵长类动物物种的CNS和外周神经系统中实现稳定转基因表达。最后,通过改变细胞特异性启动子、进一步修改衣壳变体或使用特定的AAV血清型,研究人员已经能够实现对血管周细胞、内皮细胞和星形胶质细胞的细胞选择性AAV转导。
随着这些工程策略的进行,研究正在进行中,以剖析和应对AAV载体必须传递的特定基因治疗方式所提出的挑战。对于基于CRISPR–Cas的模式,通常使用的Cas9变体占据了大约4.1–4.5 kb的DNA长度,而伴随的引导RNA占据了大约0.1 kb。因此,它们的累积大小可能会排除在标准AAV载体中包装(大约4.7 kb)。最初尝试工程更小的Cas9同源物成功地绕过了包装瓶颈,但需要更长的原间隔相邻基序(PAM)序列(参与Cas9与目标DNA的结合)并且相对于未截断形式具有更低的靶向效率。此后,设计了各种小Cas9同源物,它们具有增强的结合效率和特异性,同时适应识别多样的PAM序列。AAV大小限制也促进了AAV载体共同转导(双AAV载体)和分割货物的方法的发展。然而,值得注意的是,病毒载体长期表达Cas9从免疫角度引发了担忧,其持续的核酸酶活性可能导致随时间推移的非目标效应和意外的遗传后果。因此,可能需要使用非病毒传递系统来实现CRISPR基因编辑所需的Cas9的临时表达。
使用具有更大包装能力的病毒载体也可以扩大可以使用的操纵模式库。例如,高容量腺病毒载体(HCAds)不具有病毒编码序列。这扩大了它们的包装能力到37 kb,使它们适合携带ZFNs、TALENs和CRISPR–Cas系统。HCAds对于大型、多重化的CRISPR–Cas系统特别有益,这些系统可以同时靶向多个基因,并且可以用于多基因疾病。尽管AAVs也具有多重化潜力,但它们在安全性方面存在显著问题,特别是关于它们在Cas9核酸酶诱导DSBs后整合到大脑内的治疗基因中。以这种方式,HCAds和其他创新的病毒平台,它们能够实现高通量传递,可能会简化原本需要单独试验或在单一试验中需要多个侵入性剂量的治疗干预。
对于基因沉默模式,向CNS中的细胞群体传递足够的遗传有效载荷是一个主要挑战,因为它们的持续时间相对较短,这意味着需要重复给药。RNAi等基因沉默模式比用于基因增强或编辑的模式更小,允许它们被装载到AAVs中,这有助于提高转导效率和持续沉默。实际上,已经探索了编码RNAi的AAVs用于CNS疾病,如ALS或亨廷顿病。系统性给药融合到多丙氨酸肽(AAV-AS)的AAVs,编码针对HTT的miRNA,在小鼠CNS中实现了33–50%的基因敲低。令人印象深刻的是,AAV-AS在转导纹状体和大脑皮层的神经元方面表现出比传统使用的AAV9s更高的效率。最后,将编码针对突变SOD1的shRNA的AAV9通过脊髓和腰椎脊髓的脊髓下皮层传递给早期发病ALS的小鼠模型,延缓了疾病进展,并导致持续5个多月的神经保护效果。
正如这些例子所展示的,病毒载体的临床效用可以通过合理地将每种基因操纵模式与适当的载体配对来增强。这可能允许以最小剂量实现最大的治疗效果,因此防止未来的临床试验受到高剂量病毒基因疗法相关的致命和非致命毒性的影响。AAV介导的肝脏毒性风险是一个特别紧迫的问题,在临床试验中受到高度监控。更有效的AAV衣壳的发展可能会导致减少载体负荷,从而降低与系统性基因传递相关的毒性水平。
尽管病毒载体存在缺点,以及它们所伴随的制造和可扩展性挑战,但它们对神经细胞的可靠转导、增强的转基因表达和对工程策略的适应性或许解释了它们在大多数正在进行的临床试验中的持续使用。重要的是指出,AAV载体工程的范围远远超出了这篇综述所涵盖的内容(关于设计和特定的AAV载体工程技术的更多细节可以在最近的文献中找到)。
3.2 非病毒传递
对病毒载体的长期担忧激发了寻找替代技术的动力。非病毒载体,如化学缀合物和纳米颗粒,表现出更好的安全性、相对容易的生产、减少对环境降解的敏感性和高基因载荷效率。非病毒载体由广泛的材料组成,这些材料在它们的物理化学属性(大小、电荷、刚度和形状)以及它们能够结合促进大脑靶向配体方面提供了极大的灵活性。大小在100纳米以下的颗粒具有增强穿透BBB的能力,而结合能够针对大脑内皮细胞或神经细胞上的受体的配体可以促进细胞特异性传递。用polysorbate 80、poloxamer、转铁蛋白和谷胱甘肽等材料涂覆纳米颗粒表面进一步增强了它们穿透BBB的能力。
纳米颗粒提供的排列组合可能性已被利用来针对各种CNS病理。除了化学修饰,使用物理工具如磁转染和聚焦超声可以增强纳米颗粒对特定大脑区域的精准靶向。磁转染是一种策略,通过将遗传物质加载到磁性载体(通常是铁纳米颗粒)中,然后应用外部磁场将载体集中在体内的期望区域。另一方面,聚焦超声涉及应用声波来穿透BBB,之后非病毒载体或充满气体的微泡可以被定向到大脑的特定区域。这种方法已成功地帮助在阿尔茨海默病、帕金森病和亨廷顿病模型中非病毒传递编码神经营养因子的基因。尽管在临床前探索非病毒载体携带基因编辑工具方面的研究有限,但新兴的证据突出了它们将CRISPR–Cas9系统传递到大脑的潜力。
非病毒载体尚未在CNS基因治疗的临床试验中找到位置,可能是由于它们在有限的BBB穿透、低基因转移效率和短期基因操作方面的挑战。此外,通过静脉注射将非病毒载体传递到大脑的效率仍然不理想。例如,通过转铁蛋白受体靶向大脑的纳米颗粒通常导致不到0.1%的大脑传递效率。某些情况,如创伤性脑损伤,会诱导BBB破裂,这可以被非病毒载体利用来传递基因治疗。例如,在脑损伤后5分钟系统性给予针对神经元的纳米颗粒在小鼠受损部位附近的大脑组织中累积。然而,开发一个能够在这个破裂窗口之外传递基因治疗的系统将是至关重要的。为了实现这一点,开发了一个纳米颗粒平台,用于在创伤性脑损伤中独立于BBB病理生理学的siRNA靶向MAPT的传递。联合调节纳米颗粒的表面化学和涂层密度,无论颗粒是在破裂窗口内还是外注射,都实现了MAPT的40-50%沉默。需要更多的努力来以一致和可控的方式增强BBB穿透。
研究工作一直在持续地进行,以修改非病毒载体的可调节组分,并确定它们是否可能更适合携带某些有效载荷而不是其他。然而,需要进行广泛的筛选以确定理想的配方,这需要高通量方法来提高筛选效率。在身体的其他部位,使用DNA条形码已经使得同时在体内评估成百上千的纳米颗粒的分布成为可能。类似地,编码肽的mRNA条形码已经允许高通量筛选离子化脂质,以最大化肺部的基因表达。需要类似的努力来进行广泛的筛选,以构思一个完美的CNS载体;然而,仅通过物理化学属性的修改能否延长这些系统所关联的有限转基因表达仍然是未知的。如果解决了非病毒载体在效率低下的转染和BBB渗透性方面的问题,那么它们在可扩展性、较低的制造成本和更大的灵活性方面所提供的优势可能使它们成为受欢迎的病毒载体的替代品。
4. 临床转化的考虑
尽管有着充满希望的候选基因干预措施,但大多数针对中枢神经系统(CNS)疾病的基因治疗临床试验结果并不理想。系统地检查与传递方法、给药途径、体外和体内模型以及患者异质性相关的障碍,可以使研究者们设计更好的基因治疗和临床试验,最终提高转化成功的几率(图4)。
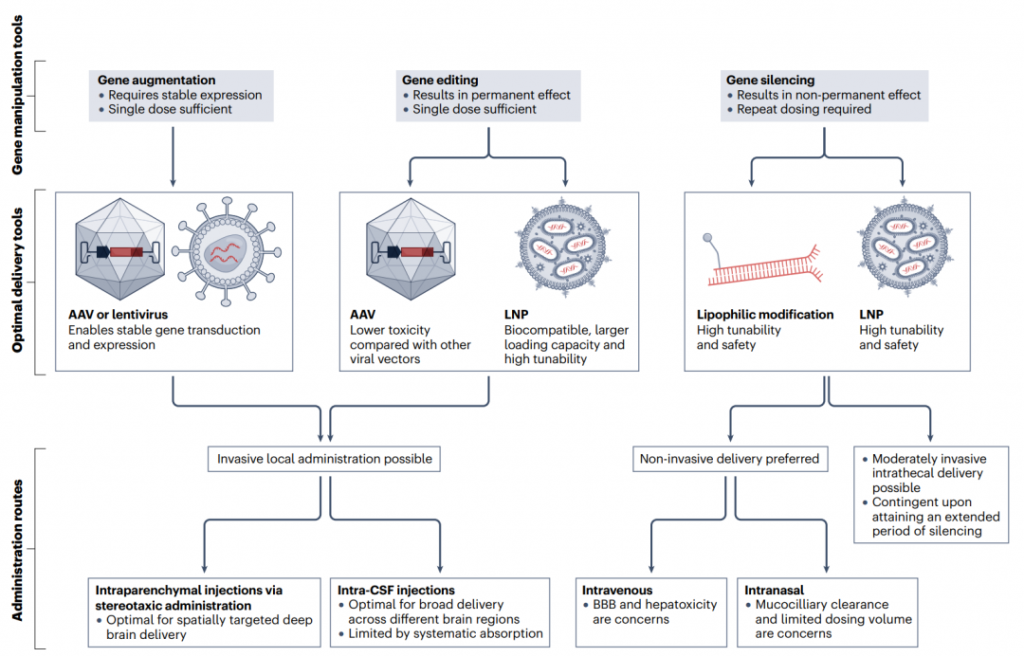
图4. 基因操作方式、传递载体和给药途径的关联性
4.1 传递系统
病毒载体在获得批准用于CNS疾病的基因治疗中占据主导地位。然而,许多挑战仍然限制了这些方法。病毒载体的可扩展性和产量差是众所周知的,一批中经常产生多个空衣壳,以及整体上多步骤的迭代制造过程增加了最终产品的成本。实际上,Skysona的每次治疗成本大约为300万美元,使其成为世界上最昂贵的治疗方法,显著阻碍了可及性,特别是对于低收入和中等收入国家的人们。在合成之外,病毒载体触发的宿主免疫反应是一个长期的安全问题,也可能导致疗效降低。这在考虑如siRNA介导的基因沉默等模式时尤其相关,这些模式很可能需要重新给药(重新给药也增加了宿主产生中和抗体的机会)。尽管在试验中已结合了免疫抑制药物,但由于记忆免疫反应和不良反应的风险,病毒载体对CNS疾病的重复给药成功仍然存疑。从失败的临床试验中学到的经验可以为未来的载体和研究设计提供关键见解;然而,许多涉及AAV载体的终止试验没有提供充分的终止理由,导致人们对原因的猜测。
非病毒传递方法在CNS疾病的应用也面临多重挑战。例如,需要精确控制脂质纳米颗粒(LNP)组分,以实现高效的细胞内传递和高转导效率。对于系统性基因治疗,BBB的穿透性和有效的大脑分布是主要障碍。LNP在肝脏中的积累也引起了肝脏毒性问题。需要核内传递的DNA疗法要求开发更先进的LNPs,这可能增加了制造过程的固有复杂性。同样,为了实现神经趋向性,将配体如肽或抗体结合到LNPs上可能会导致合成过程中的批次间变异,并进一步使转化复杂化。最后,通过非病毒传递系统传递的基因疗法的效果持续时间必须延长,以减少给药频率。
4.2 给药途径
精心选择的传递工具必须与最合适的给药途径配对,这取决于CNS疾病的时间空间表现。对于病毒载体,不同的给药途径影响其体内动力学、生物分布和免疫原性(如最近的预印本报告),以及病毒脱落(病毒粒子从体内的排出),这具有强烈的公共卫生意义。
给药途径的侵入性将决定其适用于需要重复给药策略的可行性(尽管重要的是要注意,无论给药途径如何,导致免疫反应的疗法都不太可能适用于重复给药)。静脉注射因其给药便利性和适合重复给药而具有吸引力。然而,低BBB穿透性和首过代谢可能导致低CNS转导和/或转染效率,以及增加肝脏毒性的风险。微泡增强的聚焦超声(MB-FUS)可以以时间和空间控制的方式暂时使BBB通透。例如,将MB-FUS与阳离子纳米颗粒结合使用,将siRNA传递到大脑的效率提高了十倍。
脑实质内给药经常在临床试验中被利用,因为它允许直接向目标区域传递。然而,这种途径高度侵入性,因此最适用于仅需一次性治疗的疗法(基因增强或基因编辑)。有时,脑实质内给药必须与实时磁共振成像结合使用,以提高准确性并最小化附带损伤。这种给药途径还只提供对受影响区域的有限覆盖,需要使用策略如对流增强传递来改善药物分布。对载体与细胞外基质之间相互作用的深入理解将有助于控制注射后的分布。尽管脑实质内途径的侵入性是一个问题,但应该与所针对疾病的严重性以及可能已经存在颅内干预措施的可能性(如用于治疗帕金森病运动症状的深脑刺激电极)相平衡。同样,围绕AAV介导的毒性和药物诱导的肝损伤的担忧应该与潜在的颅内手术相关的不良事件和副作用相权衡。
脑脊液内给药绕过了BBB,允许治疗更直接地传递到CNS,与系统性注射相比。由于脑脊液在CNS中循环,治疗可以传递到更广泛的区域,这可能对影响多个大脑区域的疾病有益。尽管比脑实质内给药侵入性小,但这种途径不适合需要深度脑穿透治疗的疾病,因为治疗在大脑组织中的扩散速度远慢于脑脊液推动药物清除的速率,从而最小化了载体在期望的大脑区域内的接触时间。此外,通过这条路线重复给药可能会增加脑膜炎损伤的风险并降低患者的依从性。
鼻内给药提供了一个替代的非侵入性途径来绕过BBB。通过鼻内传递的siRNA负载的PNP已成功在海马、丘脑、下丘脑和小脑中被检测到。然而,鼻内给药存在一些缺点,包括可注射体积小(限制了可以给药的总剂量)、药物的生物利用度低、给药效率取决于鼻腔通道的状况以及药物在嗅觉粘膜中的滞留。
4.3 体外和体内模型
评估基因疗法有效性的一个重要挑战是体外和体内获得的结果之间的相关性差。在各种基因治疗应用中,器官体和芯片上的器官系统的进展已显示出规避这些问题的希望。然而,这些技术仍然缺乏只有在动物模型中才能完全评估的某些生理细微差别。例如,器官体通常专注于特定组织,这使得捕捉更广泛的系统效应变得具有挑战性,而芯片上的器官系统可能无法完全复制体内观察到的免疫反应范围。
选择合适的动物模型至关重要,因为不当的选择会导致临床试验中治疗方法的效果不佳。大型动物,特别是非人灵长类动物,比啮齿类动物更好地再现人类神经生态位的结构和功能复杂性。然而,伦理问题和高成本限制了它们的可及性,使啮齿类动物成为主要选择。在推断结果之前,了解每种模型的前景和局限性至关重要。例如,尽管表达人类APP突变形式的小鼠是模拟阿尔茨海默病中神经退行性变的首要选择,但这些模型中的行为改变与淀粉样β蛋白聚集的相关性很差。人源化的小鼠模型规避了这些问题中的许多,但它们本身也很难处理。这在AAV基因治疗评估的背景下尤其如此,因为人源化的小鼠通常是免疫缺陷的,这使得研究AAV的免疫原性变得困难。基因编辑工具已经导致了更准确的动物模型的创建,并可能推动下一代模型的开发,以实现更可靠的临床前评估。同样值得注意的是,神经系统疾病的遗传表现受到年龄和性别的强烈影响,这要求在临床前研究中包含平衡的队列。
4.4 患者异质性
临床试验成功的一个关键因素是受影响个体的异质性,无论是在疾病表现还是对治疗干预的反应方面。改善患者分层的方法应该提高临床试验的成功率。例如,尽管tominersen在亨廷顿病的III期试验中被终止,但研究数据表明,该药物可能帮助较年轻且疾病较不发达的人。目前正在计划进行中期临床试验来测试这一理论。捕获推动特定疾病的全部遗传因素可以帮助更好的分层,但目前的技术还无法实现。某些变量,如BBB的渗透性和区域易感性,在个体间也存在显著差异,使得可靠测量变得困难,并可能削弱得出结论的力量。同样,关键病理事件的动态性质,这些事件在临床试验过程中可能会发生变化,需要考虑。根据疾病严重程度对患者分层进行改进可以帮助研究人员设定现实的主要和次要终点。实施更严格的纳入和排除标准可能会减少研究规模,但可能在更同质的群体中提高试验成功的机会。
对于基因治疗来说,尤其重要的是评估表观遗传和等位基因特异性对异质性的贡献和群体大小。像CRISPR这样的编辑工具可能实现等位基因特异性靶向,以实现更高的灵敏度,并引发不受导向RNA和目标DNA序列不匹配影响的编辑反应。这可能赋予靶向具有显性遗传模式或由单个核苷酸变异支撑的疾病的能力。事实上,大多数基因组变异起源于单核苷酸多态性(SNPs),过去已经通过使用siRNAs实施了等位基因特异性干预。然而,患者异质性可能影响这些潜在的等位基因特异性靶向方法,使它们难以实现。即使在明确定义的患者群体中,基因组位点的等位基因变异可能仍然难以靶向,而且采取的规避这种变异的方法可能导致应用的治疗干预和试验设计的成本更高。另一方面,作为某些单倍型的共同SNPs可以用于靶向突变等位基因,从而实现更具成本效益的等位基因特异性治疗。两项研究提出了这种策略来治疗亨廷顿病,表明基于SNP的等位基因特异性个性化治疗确实可以应用于神经退行性疾病。
最后,用于神经系统疾病的预测性、预后性和安全性生物标志物的日益增长的库也提供了进一步扩大分层程度的机会。与基因治疗相关的一个经典陷阱是背景突变对应用干预措施的解释的影响,这要求开发能够更好地预测这些非目标现象的技术。然而,从另一方面看,开发和验证能够分析非目标效应的工具可能是耗时的。此外,一旦这些工具被部署,它们必须对现有的基因治疗系统进行询问和纠正,直到检测到最小的非目标效应或没有非目标效应,所有这些都可能延长从临床前到临床研究的进展时间线。尽管如此,作者对进入一个时代持乐观态度,在这个时代,考虑到患者特定的突变、风险和结果,可以使精准的基因干预在神经疾病中得到广泛应用。
5. 总结和展望
在这篇综述中,作者阐明了各种基因治疗方式和应用于治疗CNS疾病的传递方法所提供的承诺。作者还概述了各种传递载体和给药途径的优缺点,并提出了如何将基因操作方式与传递方法和给药途径相匹配的方法。此外,作者还深入探讨了将CNS疾病的基因治疗转化为临床应用的关键挑战。基因治疗为CNS疾病提供精心设计、长期解决方案的潜力是可以实现的,只要不断适应由不同的DNA和RNA编辑方式塑造的不断发展的格局。
例如,光遗传学和化学遗传学通常不被列为本综述中概述的传统基因治疗应用的同一类别,因此在这里没有深入覆盖。然而,如设计师药物专门激活的受体(DREADDs)和药物选择性执行模块(PSAMs)等光遗传和化学遗传技术正在开创基因治疗的新时代,因为它们允许精确操纵基因定义的大脑细胞。特别是化学遗传学在各种神经发育、神经精神和神经回路功能障碍疾病的背景下显示出巨大的前景,并有可能定义下一代基因治疗。
遗传学和神经科学研究的进步不断加深对CNS疾病机制的理解。这为研究者们提供了宝贵的见解,从而识别出更有希望的治疗靶点,从而增加开发成功疗法的可能性。从传递的角度来看,BBB的复杂性质和不同CNS疾病中神经元和神经胶质景观的复杂变化提出了主要挑战。所有变量——包括免疫调节、微血管动力学和细胞特异性差异——都应该被考虑,以实现成功治疗途径的开发。从长远来看,针对个人的特定遗传档案量身定制的基因治疗方法对CNS疾病的治疗至关重要。通过考虑每个人的独特遗传变异和疾病特征,这种方法有潜力提高治疗结果并最小化非目标效应。
还需要集中努力通过非侵入性给药将基因治疗有针对性地传递到大脑的特定区域或细胞。例如,体内选择肽展示AAV库可能有助于识别能够转导CNS中的小胶质细胞的AAV衣壳变体,并随后优化以有效增加小胶质细胞趋向性和表达。基于在单个载体中表达两个或多个转基因包的组合基因治疗,也可能提高功能性效果。
在所有这些进步中,需要在创新传递工具和克服现有转化挑战之间取得平衡。还需要降低基因治疗的成本,以增强可及性,特别是对于资源有限环境中的个体。实现这一目标需要在研究开发、制造、监管流程和全球合作方面的共同努力。这些集体行动对于应对挑战和促进CNS基因治疗领域的全球健康公平至关重要。
转自:NeuExpress https://mp.weixin.qq.com/s/hW493-nN8TnHBPTIqkRhHw
